La
maladie d'Alzheimer touche environ un million de personnes en France. C'est une maladie neurodégénérative hétérogène quant à son origine, son évolution clinique ou ses lésions. Aujourd'hui, il n'existe pas de traitement curatif. La maladie est liée à la présence de dépôts de protéines amyloïde-β (Aβ) qui entraînent une cascade d'événements menant à l'accumulation de protéines tau anormales dans le cerveau, à une perte de synapses
et à terme, aux altérations de la mémoire.
Les maladies à prions, également neurodégénératives, sont causées par une protéine prion qui a changé de forme, devenant ainsi pathogène. En effet, le prion est naturellement présent dans l'organisme dans sa forme normale. Le prion pathogène peut transmettre sa forme anormale à des variantes normales du prion. Il entraîne ainsi une réaction en chaîne qui amplifie la maladie. Ce mécanisme de transmission de proche en proche par une protéine de sa forme anormale est appelé ''prion like''.
Plusieurs études ont établi que l'administration intracérébrale de composés contaminés par des protéines Aβ peut induire une pathologie amyloïde-β, selon le mécanisme ''prion-like''. Suite à cette découverte, la protéine Aβ est maintenant considérée comme une protéine pseudo-prion.
Certaines mutations génétiques de la protéine Aβ peuvent entraîner un développement précoce de la maladie d'Alzheimer. Des habitants de l'Islande ont une mutation génétique qui induit une production de formes très rares d'Aβ appelée Aβice. De façon très surprenante, ces personnes ont un vieillissement cognitif amélioré et sont protégées de la maladie d'Alzheimer. Jusqu'à maintenant, les mécanismes de cette protection étaient mal connus.
Dans leur étude publiée dans
Molecular psychiatry, des chercheurs de MIRCen, département de l'Institut Jacob ont ainsi montré qu'une seule injection intracérébrale de la forme mutée Islandaise d'Aβ à des souris modèles de la maladie d'Alzheimer les protège contre les atteintes cognitives : plusieurs mois après l'injection de Aβice, alors que ces modèles auraient dû développer la maladie d'Alzheimer, ils restent asymptomatiques. L'origine de cette amélioration est surprenante, car, au-delà des dépôts amyloïdes, l'administration d'Aβice protège contre l'apparition d'agrégats de protéines tau et réduit les altérations synaptiques ainsi que la destruction des synapses par les cellules de l'inflammation. L'étude souligne qu'un seul événement régulant l'agrégation Aβ et la santé synaptique peut protéger contre la maladie d'Alzheimer sur le long terme. En général, l'hypothèse "pseudo-prion" de la maladie d'Alzheimer suggère que les protéines Aβ peuvent amplifier un processus pathologique. Dans cette étude, les chercheurs montrent que les "pseudo-prions" peuvent également être protecteurs, ce qui ouvre la voie à la recherche d'une nouvelle catégorie de thérapies contre les maladies neurodégénératives.
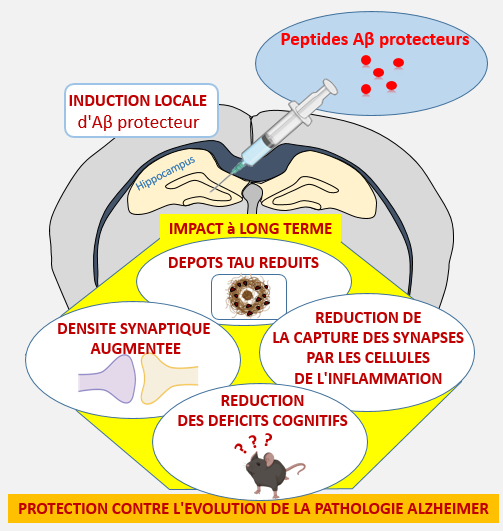
L'exposition suite à une inoculation intracérébrale de peptides Aβ protecteurs réduit la production des lésions tau. Ces améliorations sont associées à une augmentation de la densité synaptique, à une réduction de la capture des synapses par les cellules de l'inflammation et à la correction de troubles cognitifs plusieurs mois après l'inoculation. Ces données suggèrent que les mécanismes associés à cette transmission ont un rôle protecteur contre la maladie d'Alzheimer.