L’amélioration des technologies de caractérisation moléculaire à grande échelle des échantillons biologiques est à double tranchant. D’un côté, cet accès fiable et rapide à des milliers de gènes, transcripts, protéines ou métabolites offre la possibilité de vérifier un nombre considérable d’hypothèses concernant le fonctionnement du vivant. D’un autre côté, la multiplication des hypothèses étudiées simultanément augmente le risque que l’une d’entre elles soit validée par hasard et à tort (une fausse découverte). Cette augmentation est d’origine combinatoire : la probabilité est faible qu’une molécule prise au hasard subisse des fluctuations de mesures correspondant exactement aux attentes induites par l’hypothèse étudiée. En revanche, si plusieurs milliers de biomolécules sont étudiées simultanément, la probabilité qu’au moins l’une d’entre elles se comporte ainsi devient importante.
Pour contrôler le risque de fausses découvertes, des méthodes statistiques avancées sont nécessaires car les plans d’expérience deviennent de plus en plus élaborés. C’est notamment le cas en
protéomique, où la complexité de la mesure réalisée (grâce au couplage de la spectrométrie de masse et de la chromatographie liquide) vient s’ajouter au faible nombre d’échantillons qu’il est généralement possible d’analyser. Depuis de nombreuses années, des chercheurs de l’Irig travaillent donc sur l’articulation des contraintes expérimentales et des hypothèses théoriques nécessaires au contrôle des fausses découvertes, afin de proposer des chaînes d’analyses de données dotées d’un contrôle qualité rigoureux (ex :
www.prostar-proteomics.org). Leurs récents travaux se sont concentrés sur la théorie des filtres « Knockoffs » qui a révolutionné le champ de l’inférence sélective en proposant de s’appuyer sur des tirages aléatoires pour mieux caractériser les propriétés des fausses découvertes. Ils ont notamment fait le lien entre ces filtres et les méthodes empiriques de contrôle des fausses découvertes historiquement utilisées par les protéomiciens, ce qui permet de proposer de nouvelles manières de travailler [1, 2].
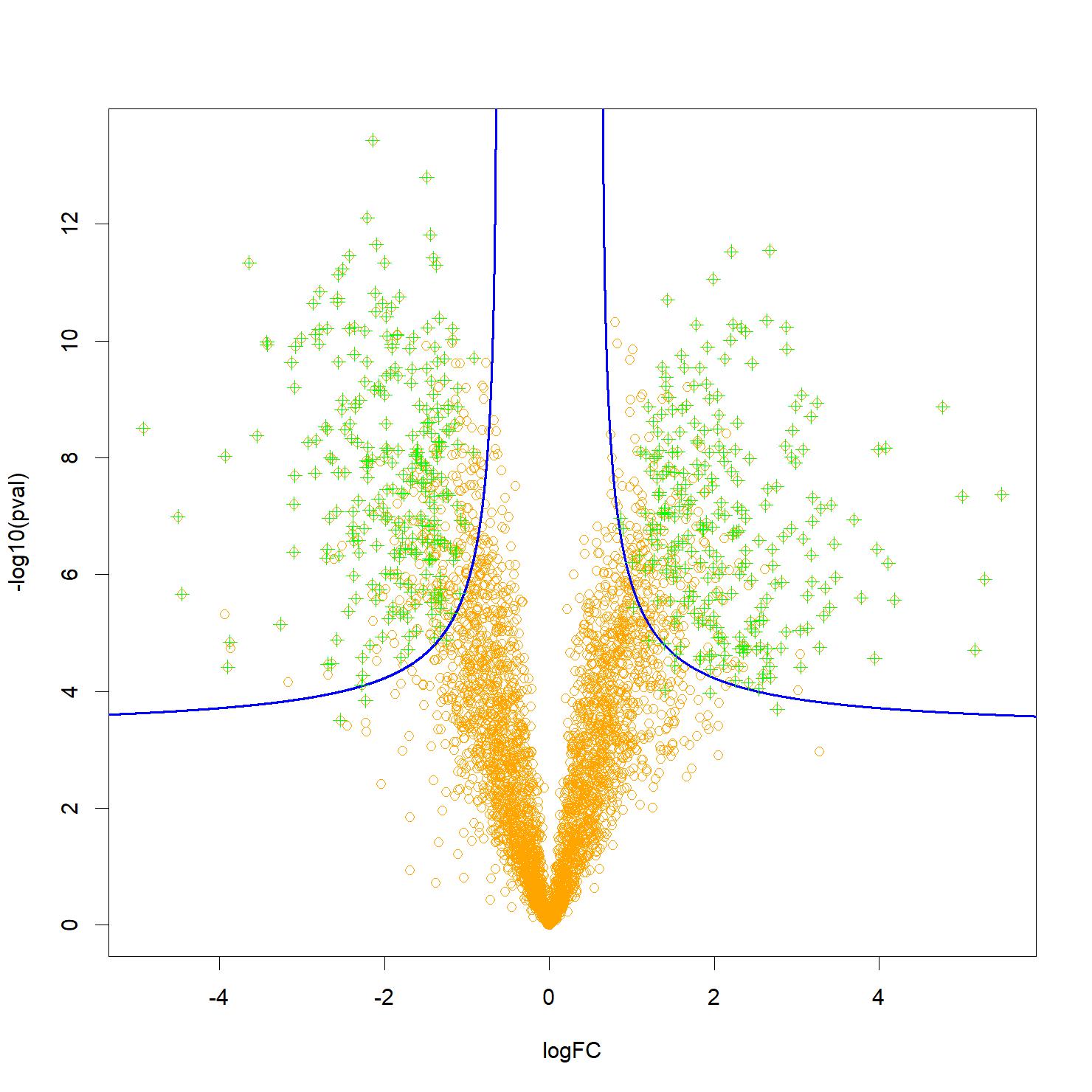
Figure : Un « volcano-plot » typique, représentant par des points oranges les protéines analysées, et pouvant potentiellement expliquer une différence de phénotype (par exemple sain ou malade) en fonction de leur significativité (en ordonnée) et de l’importance de l’effet mesuré (en abscisse). Les biomarqueurs candidats les plus pertinents sont généralement situés près des deux coins supérieurs, mais certains peuvent se trouver plus bas et au milieu, ce qui complique la sélection. Les filtres Knockoffs permettent de contrôler le taux de fausses découvertes associé à une sélection des protéines (en vert) suivant une frontière de décision plus flexible, notamment hyperbolique (représentée ici en bleu) ce qui permet de tenir compte à la fois de l’effet et de la significativité. Crédit CEA
Avec le soutien financier de l’ANR :
- Projet Multidisciplinary Institute in Artificial Intelligence (MIAI @ Grenoble Alpes)
- Le programme GRAL
via Chemistry Biology Health Graduate School at University Grenoble Alpes
- ProFI (Proteomics French Infrastructure)
Protéomique : Caractérisation (identification et quantification) à large échelle des protéines présentes dans un échantillon biologique.
Inférence sélective : Domaine des statistiques en grande dimensionnalité qui s’intéresse à la généralisation de connaissances tirées de données expérimentales alors que ces données ont été préalablement sélectionnées en raison de leurs spécificités.