Technological improvements for large-scale molecular characterization of biological samples is a double-edged sword. On the one hand, this reliable and rapid access to thousands of genes, transcripts, proteins or metabolites enable the verification of a considerable number of hypotheses about living organisms. On the other hand, the manifold of hypotheses studied simultaneously increases the risk that one of them is incorrectly validated by chance (a so-called “false discovery”). This increase roots in combinatorics: the probability is low that a random molecule displays measurement fluctuations that match to the expectations induced by the hypothesis studied. However, if several thousand of them are considered simultaneously, the probability that at least one of them behaves accordingly becomes significant.
To control for the risk of false discoveries, advanced statistical methods are needed as experimental designs become more and more elaborate. This is particularly the case in
proteomics, where the complexity of the instrumental set-up (mass spectrometry and liquid chromatography coupling) adds to the small number of samples that it is generally possible to analyze. For years, researchers at IRIG have therefore been working on articulating the experimental constraints and theoretical hypotheses necessary to control for false discoveries, in order to propose data analysis workflows with rigorous quality control properties (e.g. www.prostar-proteomics.org). Their recent work has focused on the theory of Knockoffs filters, which has revolutionized the field of
selective inference by proposing to leverage random draws to better characterize the properties of false discoveries. In particular, they made the link between these filters and the empirical methods for controlling for false discoveries that have historically been used by proteomic researchers, which makes it possible to propose innovative methods [1,
2].
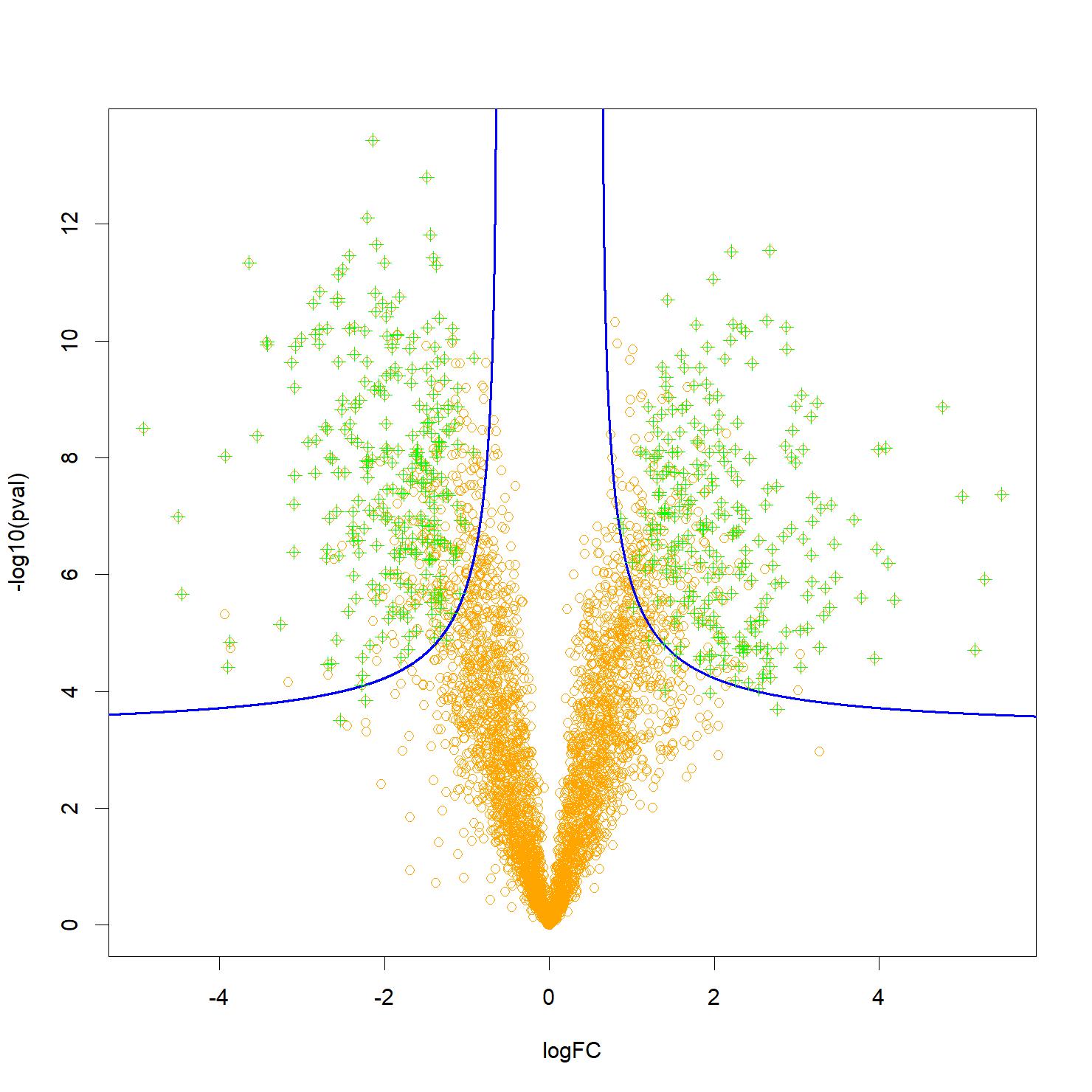
Figure: A typical “volcano-plot”, representing the proteins analyzed by orange dots, and which can explain a difference in phenotype (for example healthy or diseased), depending on their significance (on the Y-axis) and the importance of the effect measured (on the X-axis). The most relevant candidate biomarkers are usually located near the top two corners, but some may be located lower in the middle, hereby complicating selection. Knockoff filters make it possible to control for the false discovery rate associated with a selection of proteins (in green) following a more flexible decision boundary, notably hyperbolic (represented here in blue), which allows taking into account both the effect and the significance. Credit CEA
ANR Fundings
- Multidisciplinary Institute in Artificial Intelligence MIAI @ Grenoble Alpes
- Programme GRAL via Chemistry Biology Health Graduate School at University Grenoble Alpes
- ProFI Proteomics French Infrastructure
Proteomics: large-scale characterization (identification and quantification) of proteins present in a biological sample.
Selective inference: a field of high-dimensional statistics, which deals with the generalization of knowledge drawn from experimental data where the data have been previously selected for their specific characteristics.