Les équations de la mécanique quantique sont utilisées pour décrire et prédire les structures et les propriétés des molécules. Cependant, jusqu’à présent, rares étaient les méthodes capables de calculer plus de quelques centaines d’atomes. Des chercheurs de notre institut ont développé une méthode de simulation, QM-CR, qui permet de simuler des milliers d'atomes en utilisant la mécanique quantique (QM) associée à une réduction de la complexité des degrés de liberté (CR). Dans cette étude, ils sont parvenus à simuler la structure électronique d'environ 13 000 atomes pour prédire et caractériser les liaisons des variants de la protéine spike du virus SARS-CoV-2 avec le récepteur humain haACE2.
Les chercheurs [Collaboration] ont comparé quatre variants de la protéine spike : Wuhan, Omicron, et deux variants basés sur Omicron. Pour évaluer leurs liaisons avec le récepteur haACE2, les scientifiques ont pris en compte la contribution énergétique des acides aminés, et ils ont simulé une projection de l'effet de certaines mutations pour chaque acide aminé. Cette étude a permis de comprendre de façon plus détaillée comment différentes mutations affectent l'interaction entre la protéine spike et le récepteur hACE2. De plus, les prédictions des simulations ont été validées en comparant l'efficacité des variants de la protéine spike à se lier avec des cellules exprimant hACE2 (voir figure).
Ces résultats sont d’autant plus remarquables car lorsqu’ils avaient été publiés en 2021, la mutation A484K supposée être impliquée dans la liaison avec ACE2 n'avait pas encore été identifiée par les laboratoires d’épidémiologie. Il aura fallu attendre 20 mois plus tard pour que cette mutation soit effectivement observée dans le variant BA.2.86, confirmant tout l’intérêt des simulations quantiques pour obtenir des prédictions très avancées.
Ce modèle de simulation quantique QM-CR a démontré sa capacité à identifier les mutations cruciales pour les interactions intermoléculaires. Cette méthode non seulement aide à comprendre les mécanismes sous-jacents des liaisons protéiques, mais peut également guider la conception de nouveaux traitements très spécifiques. Ce travail souligne l'importance des outils de modélisation avancés dans l'étude des interactions biologiques complexes et leur potentiel pour surveiller l'évolution des virus et concevoir des vaccins plus robustes.
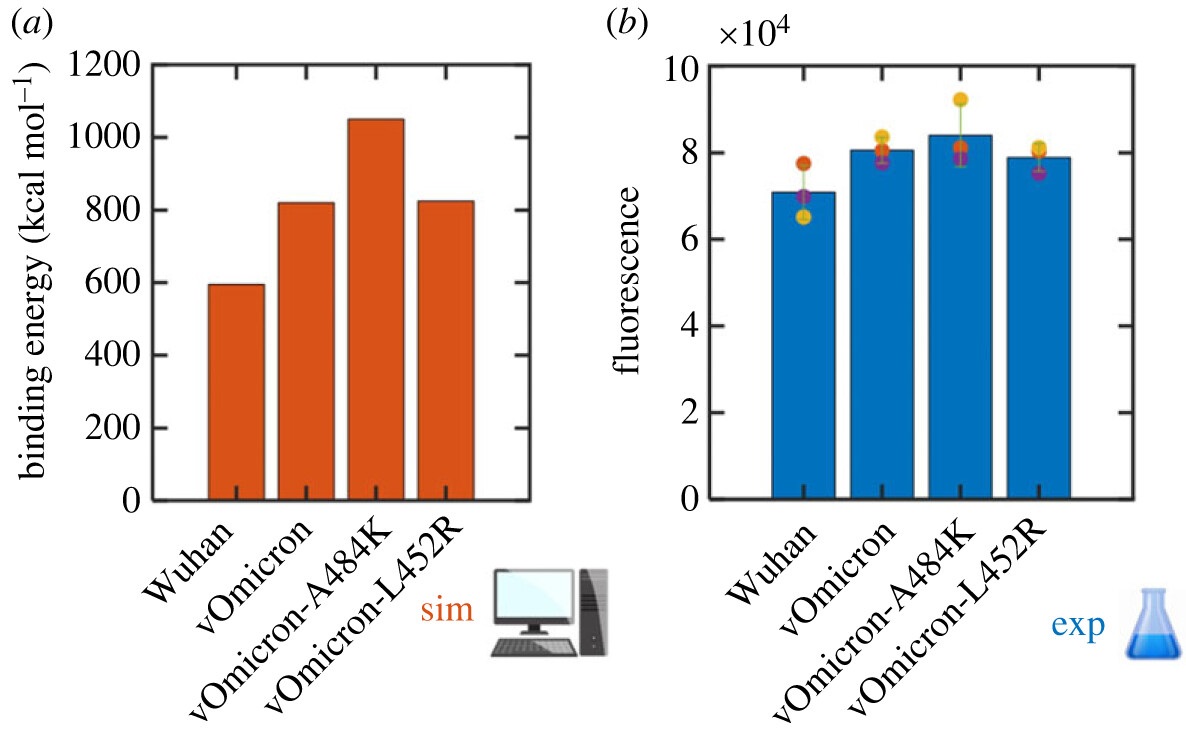
Figure : à gauche (en orange) la simulation quantique de la virulence des quatre variants du SRAS-CoV-2 est confirmée par les tests expérimentaux, à droite (en bleu). (© CEA)
Collaboration
- Boston College Department of Biology (USA)
- Harvard Medical School (USA)
- RIKEN Center for Computational Science (Japan)