The equations of quantum mechanics are used to describe and predict the structures and properties of molecules. However, until now, few methods are able to calculate more than a few hundred atoms. Researchers at our institute have developed a simulation method QM-CR, which enables thousands of atoms to be simulated using quantum mechanics (QM) combined with a reduction in the complexity of the degrees of freedom (CR). In this study, they succeeded in simulating the electronic structure of around 13,000 atoms to predict and characterize the binding of variants of the SARS-CoV-2 spike protein to the human haACE2 receptor.
The researchers [Collaboration] compared four variants of the spike protein: Wuhan, Omicron, and two variants based on Omicron. To assess their binding to the haACE2 receptor, the scientists took into account the energetic contribution of the amino acids, and simulated a projection of the effect of certain mutations for each amino acid. This study provided a more detailed understanding of how different mutations affect the interaction between the spike protein and the hACE2 receptor. In addition, the predictions of the simulations were validated by comparing the efficiency of the spike protein variants in binding to cells expressing hACE2 (see figure).
These results are remarkable because when they were published in 2021, the A484K mutation thought to be involved in binding to ACE2 had not yet been identified by epidemiology laboratories. Only 20 months later this mutation was actually observed in the BA.2.86 variant, confirming the value of quantum simulations for obtaining highly advanced predictions.
This QM-CR quantum simulation model has demonstrated its ability to identify mutations crucial to intermolecular interactions. This method not only helps to understand the underlying mechanisms of protein binding, but can also guide the design of new, highly specific treatments. This work highlights the importance of advanced modelling tools in the study of complex biological interactions and their potential for monitoring the evolution of viruses and designing more robust vaccines.
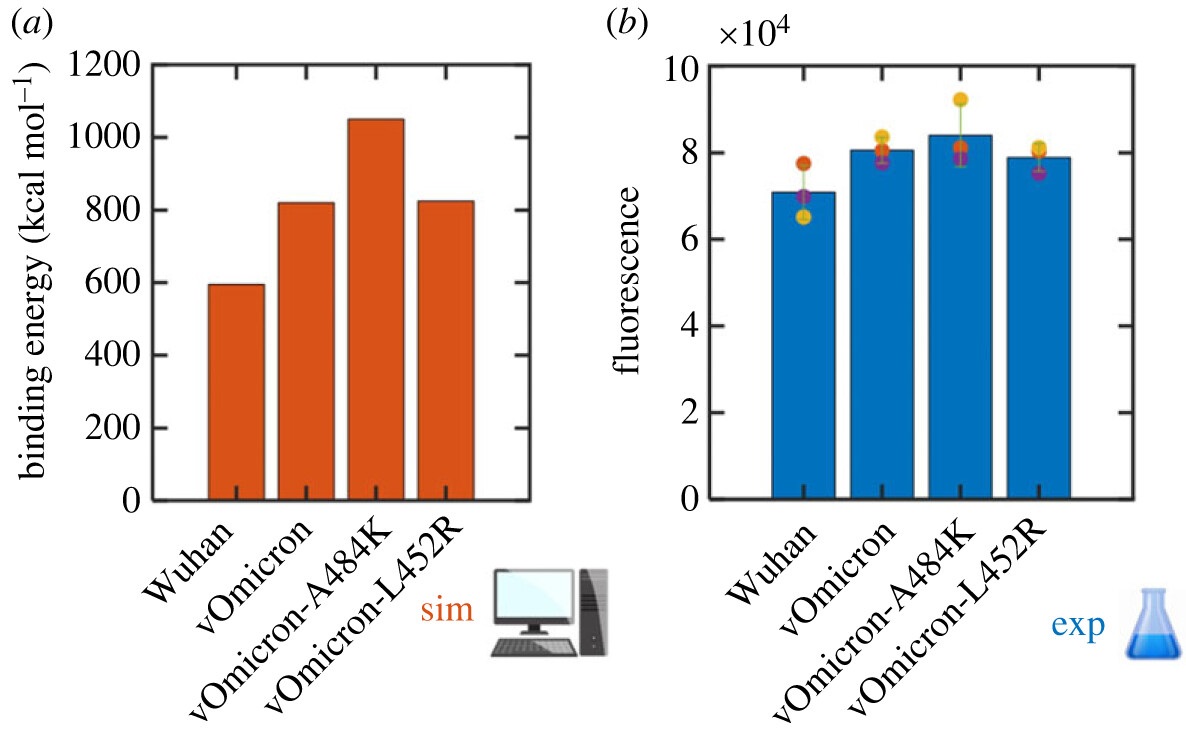
Figure: on the left (in orange) the quantum simulation of the virulence of the four SARS-CoV-2 variants is confirmed by experimental tests, on the right (in blue). (© CEA)
Collaboration
- Boston College Department of Biology (USA)
- Harvard Medical School (USA)
- RIKEN Center for Computational Science (Japan)